Risk Management Alters Medical Requirements
Many medical regulations and standards have made risk management a critical element for system and component developers. Factors like creepage and clearance are now important aspects of connector design.

Every day, medical device manufacturers get better at managing risk as they respond to international regulatory schemes that impact device design all the way down to the component level. Perhaps the most fundamental and wide-ranging of these has been the emergence and adoption of formal risk management methodologies.
ISO14971, Application of Risk Management to Medical Devices, is the international standard for applying risk management principles to the medical devices in major markets including the US, Australia, Canada, the EU, and Japan.
In the U.S., manufacturers are looking closer at components and materials, documenting risks for FDA inspection. This ISO standard has altered product selection methods and requirements for electronic components like connectors and cabling.
For years, U.S. regulations acknowledged differences in the type and level of risk represented by medical technologies. The FDA’s Center for Devices and Radiological Health shapes the regulatory processes for permitting devices to enter the marketplace in accordance their risk.
There are three risk levels, Class I, II, and III, with Class I at the lowest risk and Class III representing the highest risk level.
In the European Union, medical products are regulated through three ‘new approach’ directives that were enacted during the 1990s. To receive the CE mark, products must satisfy the ‘essential requirements’ established by the applicable directive.
Like the FDA’s classification system, the CE marking system also relies on a risk-stratified approach, applying such criteria as the duration of body contact, invasive character, use of an energy source, effect on the central circulation or nervous system, or diagnostic impact, to determine the level of risk. ISO 13485, Quality Management Systems—Requirements for Regulatory Purposes, is another important standard.
IEC 60601 also addresses medical electrical equipment. The standard has been brought into alignment with the industry’s broad regulatory trends to a risk management approach.
Manufacturers will experience this shift in these ways:
- A product’s essential performance characteristics must be examined in relation to criteria needed to avoid unacceptable risk.
- The manufacturer’s process for complying with IEC 60601-1 must itself be in compliance with ISO 14971.
- A risk management file will be required for every product. The 3rd edition of IEC 60601-1 also revises technical requirements in areas ranging from mechanical hazards such as crushing or instability to the application of alarm systems.
Impact on connectors
Virtually every one of the standard’s sections incorporates subsections that have special importance for the field of medical connectors. An example of the standard’s new and important technical requirements for connectors can be seen if we drill down even further.
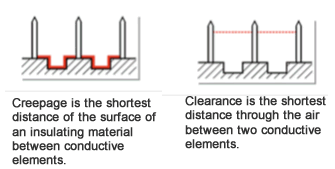
Illustrated here are the standard’s definitions and new requirements for creepage distance and air clearance, which are essential for distinguishing the means of patient protection (MOPP) from the means of operator protection (MOOP) required for a particular device.
To comply with the requirements of the new edition of IEC 60601-1, manufacturers will need to assess how creepage and air clearance in their connectors affects the risks associated with their products. To accomplish that, developers need to know how creepage and air clearance are measured.
Shown here are a couple examples from the IEC rules that are used to clarify the way that creepage and air clearance are measured under the new requirements of IEC 60601-1 (3rd ed.).
In today’s increasingly IT-enabled and interoperable world of medical devices, electrical connectors are playing a vital role in advancing product capabilities. They are also an important element that must be addressed whenever a manufacturer develops a risk assessment profile for a new or updated device.
Worldwide trends in medical device regulation favor the convergence of risk stratification, quality systems, and risk management approaches. Manufacturers will bear responsibility for conducting risk management processes in compliance with applicable standards. Working with qualified suppliers will facilitate manufacturers’ compliance with quality systems and risk management requirements.
Patrick Kinyanju is a senior engineer at Fischer Connectors, Inc.
You might like:




