EU MDR: Update to Medical Device Regulations in Europe
New regulations impact the more than 500,000 types of medical devices and in vitro diagnostic (IVD) medical devices in Europe. What do these regulations mean for device manufacturers?
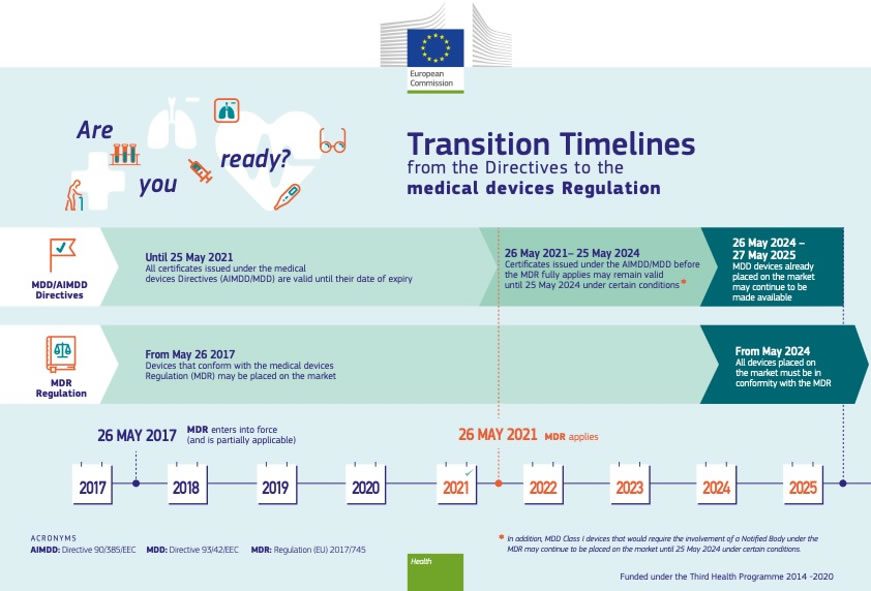
European Union Medical Devices Regulation (2017/745/ EU) (MDR) represents a major update to the Medical Devices Directive (93/42/EEC) (MDD) and the Active Implantable Medical Devices Directive (90/385/EEC) (AIMDD) from the 1990s. EU MDR was adopted in April 2017, the start of the transition period that ended with the date of application, May 26, 2021. (EU IVDR, the new rules for in vitro diagnostic medical devices, took effect May 2022.)
According to the European Commission, the motivation for these extensive updates stemmed from issues with medical devices, such as metal hips, that negatively impacted confidence in the safety of medical devices among patients, consumers, and healthcare professionals. The new regulations represent tighter controls to ensure safety and effectiveness while cultivating innovation and promoting competitiveness in the medical device sector. EU MDR more accurately reflects recent scientific and technological progress, setting the gold standard for medical device regulation. The gradual transition was implemented to give manufacturers enough time to make the necessary changes.
Some important differences in the MDR, compared to the prior directives, impact medical device manufacturers. These affect all device manufacturers supplying products to the European market, regardless of where the devices are made:
— More stringent requirements for the designation of Notified Bodies (organizations responsible for assessing the conformity of certain products before they are placed on the market), with increased control and monitoring by the national competent authorities and the Commission.
— Reclassification of certain devices with an increased scope. The MDR explicitly covers all devices for cleaning, sterilizing, or disinfecting other medical devices (Article 2.1); reprocessed single-use medical devices (Article 17); and certain devices with no intended medical purpose (Annex XVI), for example.
— Implementation of a Unique Device Identification system (Article 27) to significantly enhance the traceability and the effectiveness of post-market safety-related activities.
— Increased transparency making information on devices and studies public. The new European Database for Medical Devices – EUDAMED –plays a central role in making data available and increasing both the quantity and quality of data (Article 33).
EU MDR states the specific obligations of manufacturers. Medical device manufacturers are required to have systems for risk management and quality management, to conduct clinical evaluations, to compile technical documentation, and to apply a conformity assessment procedure. The responsibility of the manufacturers continues after their devices are on the market. This includes means to cover their financial responsibility if their device causes harm due to being defective, naming a person responsible for regulatory compliance, and providing implant cards to patients who receive certain implantable devices.
When all obligations have been completed, manufacturers must draw up a declaration of conformity and apply CE marking to their devices. Non-EU/EEA manufacturers must have a contract with an authorized representative whose place of business is in an EU/EEA member state.
Due to COVID, the original 2020 date of application was delayed one year, however, many manufacturers are still struggling to complete the regulatory process, which can take a year or more. The grace period is scheduled to end in 2024. The release of the EUDAMED database, consisting of six modules, has also been pushed to the second quarter of 2024.
- Three Companies, One Mission: Reduce Plastics in Electronic Connectors - July 28, 2026
- Cable Accessories and Tools Product Roundup - July 28, 2026
- Extreme Temperature Innovations for Aerospace and Defense - July 21, 2026